
INTRODUCTION
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is the most common enzyme defect in the world, affecting more than 350 million people. 1 The enzyme is responsible for the first step of the pentose phosphate pathway, in which NADPH is produced; hence G6PD is an essential enzyme in red blood cell for defence against oxidative stress. 1
G6PD deficiency can cause neonatal hyperbilirubinemia and chronic haemolytic anaemia, however, most of the individuals are generally asymptomatic, only experiencing episodic acute haemolytic anaemia (AHA) in the contexts of oxidative stress, such as with certain drugs or infection. 2
The G6PD gene is localized on the X chromosome (Xq28), spanning over 18 kb, and is composed of 13 exons and 12 introns. Until now, almost 400 allelic variants had been reported. 3 Patients with chronic nonspherocytic haemolytic anaemia (CNSHA), the severe type of G6PD deficiency, present mutations (Class I variants), most of them located in exons 10 or 11, that encode aminoacids located at the dimer interface, and severely affect protein stability. 3
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is the most common enzyme defect in the world, affecting more than 350 million people. 1 The enzyme is responsible for the first step of the pentose phosphate pathway, in which NADPH is produced; hence G6PD is an essential enzyme in red blood cell for defence against oxidative stress. 1
G6PD deficiency can cause neonatal hyperbilirubinemia and chronic haemolytic anaemia, however, most of the individuals are generally asymptomatic, only experiencing episodic acute haemolytic anaemia (AHA) in the contexts of oxidative stress, such as with certain drugs or infection. 2
The G6PD gene is localized on the X chromosome (Xq28), spanning over 18 kb, and is composed of 13 exons and 12 introns. Until now, almost 400 allelic variants had been reported. 3 Patients with chronic nonspherocytic haemolytic anaemia (CNSHA), the severe type of G6PD deficiency, present mutations (Class I variants), most of them located in exons 10 or 11, that encode aminoacids located at the dimer interface, and severely affect protein stability. 3
OBJECTIVE
To analyse the molecular heterogeneity of G6PD deficiency in Portugal.
To analyse the molecular heterogeneity of G6PD deficiency in Portugal.
METHODS
A total of 143 Portuguese G6PD deficient patients (104 males, 36 heterozygous females and 3 homozygous females), without known black ancestry, had been enrolled in molecular diagnosis of G6PD deficiency at the Hematology Unit from Centro Hospitalar e Universitário de Coimbra (CHUC), between 1994 and 2020. Individuals were mainly from Central Portugal, but also from Southern, Northern and Azores regions.
Diagnostic was made based on the clinical history, hematologic data and demonstration of a reduced erythrocyte G6PD activity. Enzyme quantification was addressed by quantitative spectrophotometric enzymatic assay.
Molecular study was made by PCR-RFLP analysis or by direct Sanger sequencing of all the exons and adjacent intronic regions of the G6PD gene.
A total of 143 Portuguese G6PD deficient patients (104 males, 36 heterozygous females and 3 homozygous females), without known black ancestry, had been enrolled in molecular diagnosis of G6PD deficiency at the Hematology Unit from Centro Hospitalar e Universitário de Coimbra (CHUC), between 1994 and 2020. Individuals were mainly from Central Portugal, but also from Southern, Northern and Azores regions.
Diagnostic was made based on the clinical history, hematologic data and demonstration of a reduced erythrocyte G6PD activity. Enzyme quantification was addressed by quantitative spectrophotometric enzymatic assay.
Molecular study was made by PCR-RFLP analysis or by direct Sanger sequencing of all the exons and adjacent intronic regions of the G6PD gene.
RESULTS
Mutations identified in the Portuguese G6PD deficient patients or heterozygous women are detailed in Table 1.
Table 1. G6PD pathogenic mutations found in Portuguese patients or heterozygous individuals.
Mutations identified in the Portuguese G6PD deficient patients or heterozygous women are detailed in Table 1.
Table 1. G6PD pathogenic mutations found in Portuguese patients or heterozygous individuals.
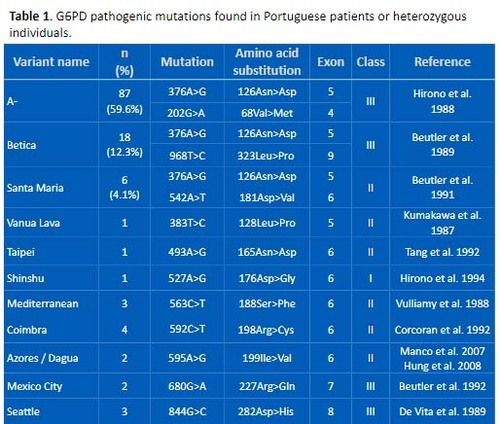
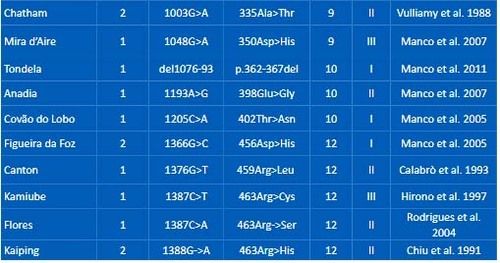
Twenty-one different G6PD pathogenic mutations were found. Among them, 20 were missense and one was an in-frame deletion of 18 nucleotides in exon 10 (G6PD Tondela). All these mutations were previously reported for the Portuguese population or other populations worldwide. 3
RESULTS (CONT)
The three most frequent mutations belong to the G6PD A (376G) African background haplotype, namely G6PD A- (202G>A) (59.6%), G6PD Betica (968T>C) (12.3%) and G6PD Santamaria (542A>T) (4.1%). Despite the African background haplotype, most hemizygous patients have non-African Ychromosomal lineages (data not shown).
From the 18 rare mutations, the most common was G6PD Coimbra, found in 4 chromosomes (2.74%). Four were described as class I G6PD mutations (Shinshu, Tondela, Covão do Lobo and Figueira da Foz). The other G6PD rare mutations present in 1, 2 or 3 individuals were: Vanua Lava, Taipei, Mediterranean, Azores, Mexico City, Seattle, Chatham, Mira d’Aire, Anadia, Canton, Kamiube, Flores and Kaiping.
Five G6PD deficient hemizygous individuals (G6PD activity at about 80% in accordance with class III G6PD mutations) have the silent mutation 1311C>T (437Tyr=) in combination with the IVS11+93C>T polymorphism, with no other mutation within G6PD gene.
Six of the rare G6PD mutations were until now only found in the Portuguese population (G6PD Tondela, Anadia, Flores, Azores, Covão do Lobo and Figueira da Foz).
The three most frequent mutations belong to the G6PD A (376G) African background haplotype, namely G6PD A- (202G>A) (59.6%), G6PD Betica (968T>C) (12.3%) and G6PD Santamaria (542A>T) (4.1%). Despite the African background haplotype, most hemizygous patients have non-African Ychromosomal lineages (data not shown).
From the 18 rare mutations, the most common was G6PD Coimbra, found in 4 chromosomes (2.74%). Four were described as class I G6PD mutations (Shinshu, Tondela, Covão do Lobo and Figueira da Foz). The other G6PD rare mutations present in 1, 2 or 3 individuals were: Vanua Lava, Taipei, Mediterranean, Azores, Mexico City, Seattle, Chatham, Mira d’Aire, Anadia, Canton, Kamiube, Flores and Kaiping.
Five G6PD deficient hemizygous individuals (G6PD activity at about 80% in accordance with class III G6PD mutations) have the silent mutation 1311C>T (437Tyr=) in combination with the IVS11+93C>T polymorphism, with no other mutation within G6PD gene.
Six of the rare G6PD mutations were until now only found in the Portuguese population (G6PD Tondela, Anadia, Flores, Azores, Covão do Lobo and Figueira da Foz).
CONCLUSION
This report confirms the wide genetic heterogeneity of G6PD deficiency in the Portuguese population.
This report confirms the wide genetic heterogeneity of G6PD deficiency in the Portuguese population.
ACKNOWLEDGEMENTS
This study was supported by CIAS / Fundação para a Ciência e a Tecnologia (FCT) [UIDB/00283/2020] and Forum Hematológico (CHUC).
This study was supported by CIAS / Fundação para a Ciência e a Tecnologia (FCT) [UIDB/00283/2020] and Forum Hematológico (CHUC).
REFERENCES
1. Luzzatto L et al. Glucose-6-Phosphate Dehydrogenase Deficiency. Hematol Oncol Clin N Am 2016;30:373–393.
2. Mason et al. G6PD deficiency: the genotype-phenotype association. Blood Reviews 2007;21:267-283.
3. Minucci A et al. Glucose-6-phosphate dehydrogenase (G6PD) mutations database: review of the "old" and update of the new mutations. Blood Cells Mol Dis. 2012;48:154-65.
1. Luzzatto L et al. Glucose-6-Phosphate Dehydrogenase Deficiency. Hematol Oncol Clin N Am 2016;30:373–393.
2. Mason et al. G6PD deficiency: the genotype-phenotype association. Blood Reviews 2007;21:267-283.
3. Minucci A et al. Glucose-6-phosphate dehydrogenase (G6PD) mutations database: review of the "old" and update of the new mutations. Blood Cells Mol Dis. 2012;48:154-65.
The data in this poster was presented at EHA Meeting 2021. Published with permission from the Copyright owner.