
ABSTRACT
It has been demonstrated that the combination of t-NGS and RBC deformability measured by OGE using a laser-assisted optical rotational ektacytometer (LoRRca) is the most powerful contribution to the clear differentiation between HS and HE. The possibility that in HPP, RBCs appear to have a significantly decreased ability to maintain deformability in the face of hypertonicity makes the classical trapezoidal profile of HE is less evident. Our objective is to provide evidence that in Hereditary Pyropoikilocytosis (HPP), the implication of a canonical splicing site, may explain, not only the marked defect of spectrin chains tetramerisation and elliptocytosis, but also the weakness of skeleton attachment to the lipid bilayer and the loss of RBC membrane with microvesiculation and formation of spherocytes as in HS.
It has been demonstrated that the combination of t-NGS and RBC deformability measured by OGE using a laser-assisted optical rotational ektacytometer (LoRRca) is the most powerful contribution to the clear differentiation between HS and HE. The possibility that in HPP, RBCs appear to have a significantly decreased ability to maintain deformability in the face of hypertonicity makes the classical trapezoidal profile of HE is less evident. Our objective is to provide evidence that in Hereditary Pyropoikilocytosis (HPP), the implication of a canonical splicing site, may explain, not only the marked defect of spectrin chains tetramerisation and elliptocytosis, but also the weakness of skeleton attachment to the lipid bilayer and the loss of RBC membrane with microvesiculation and formation of spherocytes as in HS.
INTRODUCTION
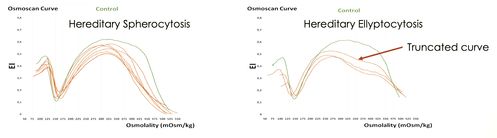
RBC morphology is a key diagnostic feature for hereditary spherocytosis (HS) and hereditary elliptocytosis (HE). However, this phenotypic diagnosis may not be readily available in patients with high reticulocyte counts and/or requiring frequent transfusions and does not allow to predict the disease clinical course or its severity. However, RBC deformability measured by osmotic gradient ektacytometry (OGE) allows a clear differentiation between HS and HE, where the truncated osmoscan curve reflects the inability of the already elliptical cells to deform further under shear stress. In the severe clinical form of HE called Hereditary Pyropoikilocytosis (HPP), the RBCs appear to have a significantly decreased ability to maintain deformability in the face of hypertonicity and the classical trapezoidal profile of HE is less evident making it indistinguishable from the HS profile. Here, two unrelated patients with severe HHA due to HPP and HS, are described with the joint inheritance of a complex set of five variants, all located in the SPTA1 gene. Two of these variants are novel and not described before, one is a microdeletion that removes the entire SPTA1 gene, and two are the known low‐expression polymorphic alleles: α-LELY and α-LEPRA. Both patients exhibit the same HS osmoscan profile suggesting that in HPP patient with marked spherocytosis, the interactions between the two SPTA1 gene variants may led , in addition to the elongation effect (elliptocytes) to a loss of membrane stability and microvesiculation (spherocytes) (1,2,3) Accordingly the RBCs appear to have a significantly decreased ability to maintain deformability in hypotonic conditions and due to this, the trapezoidal profile of HE may become indistinguishable from HS, highlighting the importance of OGE (ektacytometry) for the differential diagnosis between HS and HPP in patients with no family history of RBC cytoskeleton.
RBC morphology is a key diagnostic feature for hereditary spherocytosis (HS) and hereditary elliptocytosis (HE). However, this phenotypic diagnosis may not be readily available in patients with high reticulocyte counts and/or requiring frequent transfusions and does not allow to predict the disease clinical course or its severity. However, RBC deformability measured by osmotic gradient ektacytometry (OGE) allows a clear differentiation between HS and HE, where the truncated osmoscan curve reflects the inability of the already elliptical cells to deform further under shear stress. In the severe clinical form of HE called Hereditary Pyropoikilocytosis (HPP), the RBCs appear to have a significantly decreased ability to maintain deformability in the face of hypertonicity and the classical trapezoidal profile of HE is less evident making it indistinguishable from the HS profile. Here, two unrelated patients with severe HHA due to HPP and HS, are described with the joint inheritance of a complex set of five variants, all located in the SPTA1 gene. Two of these variants are novel and not described before, one is a microdeletion that removes the entire SPTA1 gene, and two are the known low‐expression polymorphic alleles: α-LELY and α-LEPRA. Both patients exhibit the same HS osmoscan profile suggesting that in HPP patient with marked spherocytosis, the interactions between the two SPTA1 gene variants may led , in addition to the elongation effect (elliptocytes) to a loss of membrane stability and microvesiculation (spherocytes) (1,2,3) Accordingly the RBCs appear to have a significantly decreased ability to maintain deformability in hypotonic conditions and due to this, the trapezoidal profile of HE may become indistinguishable from HS, highlighting the importance of OGE (ektacytometry) for the differential diagnosis between HS and HPP in patients with no family history of RBC cytoskeleton.
MATERIALS AND METHODS
The diagnosis of Hereditary Hemolytic Anemia (HHA) was performed via a step-wiseprocess including RBC morphology, Hb electrophoresis, and measurement of common RBC enzyme activities. RBC deformability and other rheological parameters were studied by OGE, using the osmoscan module of the Laser-assisted Optical Rotational DeformabilityCell Analyser (LoRRca; MaxSis. RR Mechatronics) as previously described (4).
The diagnosis of Hereditary Hemolytic Anemia (HHA) was performed via a step-wiseprocess including RBC morphology, Hb electrophoresis, and measurement of common RBC enzyme activities. RBC deformability and other rheological parameters were studied by OGE, using the osmoscan module of the Laser-assisted Optical Rotational DeformabilityCell Analyser (LoRRca; MaxSis. RR Mechatronics) as previously described (4).

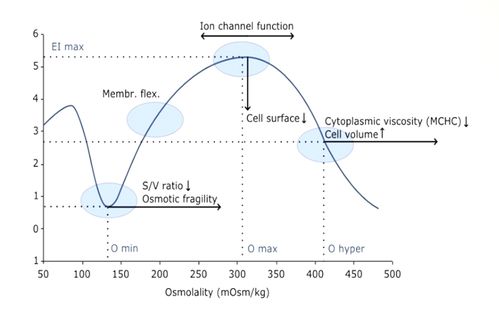
(Osmoscan): The RBCs are submitted to an increasing osmotic gradient (80 -550 mOsmol/L) under a constant shear force (<30 Pa)
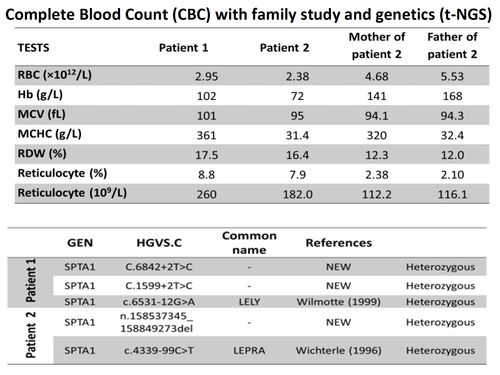
Genetic diagnosis of membranopathies was performed by gene capture followed by t –NGS that includes a panel of 35 genes responsible for membranopathies hemoglobinopathies, enzymopathies and congenital dyserythropoietic anemias (CDA) t-NGS and Whole exome sequencing (WES) have been performed usingan Illumina HiSeq2000 device.
Genetic diagnosis of membranopathies was performed by gene capture followed by t –NGS that includes a panel of 35 genes responsible for membranopathies hemoglobinopathies, enzymopathies and congenital dyserythropoietic anemias (CDA) t-NGS and Whole exome sequencing (WES) have been performed usingan Illumina HiSeq2000 device.
RESULTS AND DISCUSSION
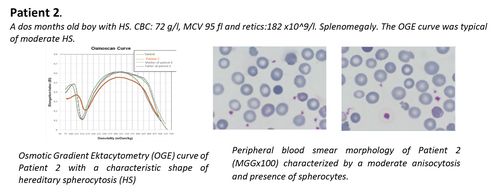
CONCLUSION
Our study demonstrates that
Our study demonstrates that
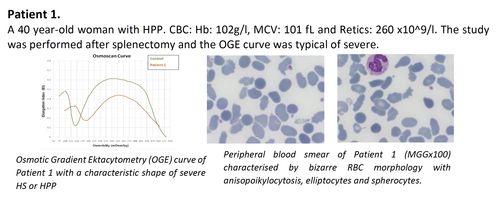
- in HPP, with a high percentage of spherocytes and typical HS osmoscan profile, the interactions between both SPTA1 gene variants, in addition to the loss of shape recovery after elongation (elliptocytes), gives rise to a membrane instability leading to spherocytes which percentage may be depending on the severity of the tetramerisation failure.
- The importance of OGE combined with t-NGS for the diagnosis of patients with RBC morphology characteristic of HPP and clinical phenotype of hereditary hemolytic anemia (HHA) of unknown etiology.
REFERENCES
1.Wilmotte R, Marechal J, Delaunay J. Mutation at position -12 of intron 45 (c-->t) plays a prevalent role in the partial skipping of exon 46 from the transcript of allele alphaLELY in erythroid cells. Br J Haematol. 1999; 104 :855-9. 2.Wichterle H. et al. Combination of two mutant alpha spectrin alleles underlies a severe spherocytic hemolytic anemia, The J. Clin.Invest.1996; 98: 2300-2307 3.Gallagher P. G. et al. Aberrant splicing contributes to severe α-spectrin–linked congenital hemolytic anemia //The J. Clin. Invest. 2019; 129: 2878-2887. 4.Llaudet-Planas E; Vives-Corrons JL; Rizzuto V; Gómez-Ramírez P; Sevilla Navarro J; Coll Sibina MT; et al. Osmotic gradient ektacytometry: A valuable screening test for hereditary spherocytosis and other red blood cell membrane disorders. Int. J. Lab. Hem. 2018; 40: 94-102
1.Wilmotte R, Marechal J, Delaunay J. Mutation at position -12 of intron 45 (c-->t) plays a prevalent role in the partial skipping of exon 46 from the transcript of allele alphaLELY in erythroid cells. Br J Haematol. 1999; 104 :855-9. 2.Wichterle H. et al. Combination of two mutant alpha spectrin alleles underlies a severe spherocytic hemolytic anemia, The J. Clin.Invest.1996; 98: 2300-2307 3.Gallagher P. G. et al. Aberrant splicing contributes to severe α-spectrin–linked congenital hemolytic anemia //The J. Clin. Invest. 2019; 129: 2878-2887. 4.Llaudet-Planas E; Vives-Corrons JL; Rizzuto V; Gómez-Ramírez P; Sevilla Navarro J; Coll Sibina MT; et al. Osmotic gradient ektacytometry: A valuable screening test for hereditary spherocytosis and other red blood cell membrane disorders. Int. J. Lab. Hem. 2018; 40: 94-102
The data in this poster was presented at EHA 2021. Published with permission from the Copyright owner.